Mass spectrometry (MS) has emerged as a core analytical technique in protein chemistry. Driven by the rapid development of instrumentation, analysis methods and computing tools, MS based proteomics is at the forefront of techniques in modern life science research and nowadays allows the study of nearly complete proteomes.
In the Mechtler lab, we are interested in developing new methods to increase sensitivity in protein identification, accuracy and precision of protein quantification as well as for the detection of posttranslational modifications. Additionally, we establish methods in the field of protein cross-linking mass spectrometry and single cell proteomics. Another focus is the development of new algorithms for MS data interpretation and bioinformatic analysis.
To achieve these goals, we frequently collaborate with chromatography and MS companies to test newest column materials, instrumentation and software.
Computational proteomics
Current MS instruments produce vast amounts of data. Therefore, automated data processing pipelines are required. Modern pipelines need to fulfill tasks ranging from peptide and protein identification, quantification and downstream statistical and bioinformatics analysis. These general tasks need to be addressed in a variety of technological and experimental setups. Furthermore, reliable operation requires tools for monitoring quality control and sample administration.
Using Proteome Discoverer (PD, Thermo Scientific) as framework we have developed several algorithms and integrated them into PD as nodes, such as the database search engine MS Amanda (Dorfer et al., J Proteome Res. 2014), ptmRS (Taus et al, J Proteome Res. 2011) for localization of post-translational modifications, apQuant (Doblmann et al., J Proteome Res. 2019) and Hyperplex for quantification of label-free and isobarically labelled samples. Nodes for normalization and statistical analysis based on limma (Smyth, Stat. App. in Genetics and Mol. Biology 2004) were also implemented and MS2Go allows to export MS results in a user-friendly format.
For quality control, we developed SimpatiQCo (Pichler et al., J Proteome Res. 2012), which allows user-friendly monitoring of key parameters of all LC-MS platforms to assure reproducible and stable performance of the systems at the highest possible sensitivity
These tools are constantly adapted to new instrument technologies and finetuned by including additional parameters.

Cross-linking Mass Spectrometry
Chemical cross-linking in combination with mass spectrometry (XL-MS) is being increasingly used in proteomics research to map sites of protein-protein interaction, gain low resolution structural information or to monitor conformational changes of protein complexes. In a cross-linking experiment, a reagent forms covalent bonds between specific residues that are in close spatial proximity to each other, thus revealing distance restraints between residues and hence interaction sites either within a protein or between different proteins. XL-MS has the unique ability to capture protein dynamics in situ, which renders it appropriate to study a range of situations where no other structural biology technique (e.g. x-ray crystallography) is applicable. In addition, XL-MS is a very sensitive technique that can be applied to endogenous levels of proteins, and proteins within complex mixtures.
We have set up methods for cross-linking protein complexes with a low number of subunits/binding partners by different cross-linkers including an enrichment procedure (Rampler et al., J Proteome Res 2015, Matzinger et al., J. Proteome Res. 2020). A synthetic peptide library of cross-linked peptides was constructed to benchmark different XL-MS software tools (Beveridge et al., Nat. Commun. 2020). Currently, we focus on the establishment of new workflows for affinity-enrichable cross-linkers with reactive sides towards different amino acids.

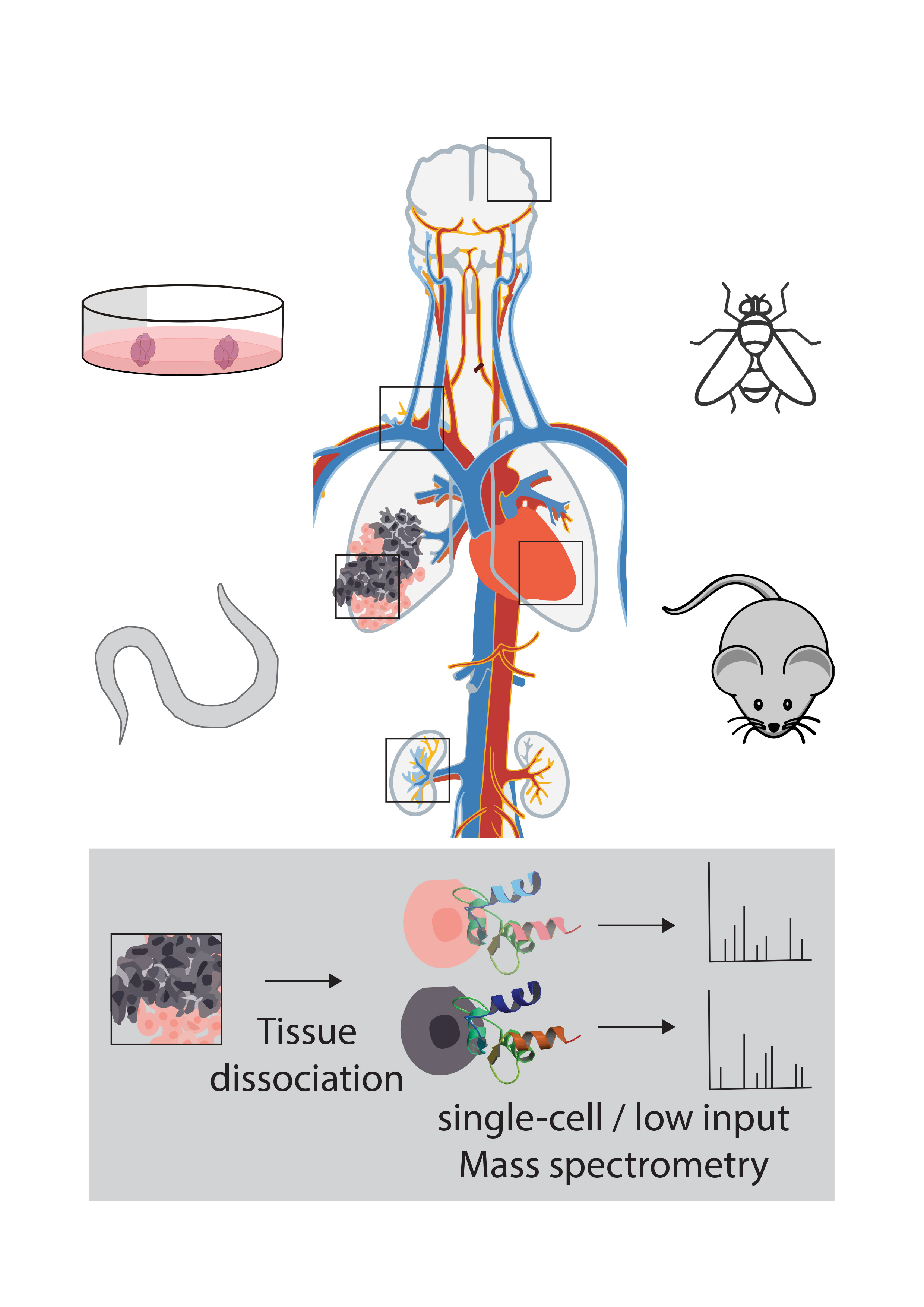
SINGLE CELL PROTEOMICS (LOW INPUT PROTEOMICS)
The rapidly emerging field of single-cell proteomics aims at complementing transcriptome and genome data by generating comparative protein expression profiles from individual cells. Despite the tremendous technological improvements in ultrasensitive proteomics workflows and instrumentation, the comprehensive characterization of individual mammalian cells still challenges the sensitivity of currently available tandem mass-spectrometry-based proteome analysis methods.
We currently aim at optimizing miniaturized sample preparation workflows by employing and further adapting published techniques for various biological systems (organoids, tissue samples) and applications (complementing sequencing techniques with proteome expression data). Furthermore, we investigate alternative labeling and acquisition strategies to enable accurate single cell or other very low input proteome measurements. These efforts involve a combination of new instruments (Bruker timsTOF Pro, Thermo Scientific Orbitrap Exploris 480) and new chromatographic systems such as the highly sensitive uPACTM columns (Stadlmann et al., Anal. Chem 2019).
Contact

Karl Mechtler