
In our studies, we combine in vivo and in vitro approaches by using embryonic stem cells and early mouse embryos. The reduced complexity of this model facilitates causal studies and provides clear phenotypic outcomes.

In mammals, development begins when an oocyte is fertilized by a sperm, forming a 1-cell embryo called a zygote. The maternal and paternal genomes remain separate and interminglefor the first time during the first mitotic division, one day post fertilization, when the 2-cell embryo is formed.
After several cleavage divisions, the embryo reaches the blastocyst stage by embryonic day 3.5 (E3.5). The main morphological differences among embryonic cells emerge at the 8-cell stage (E2), and by E3.5 two distinct lineages are specified: the inner cell mass (ICM) which gives rise to the embryo and the trophectoderm (TE) which forms the placenta.
One of the first molecular milestones, namely embryonic genome activation (EGA), occurs at the 2-cell stage, where the embryo transitions from relying on maternal transcripts to transcribing its own genome. In the following two days, lineage-specific transcription patterns arise, and 3D genome is reorganized. The first lineage decision between ICM and TE is critical not only for determining cell fate but also for restricting cell potency. Zygotes and early blastomeres are totipotent, meaning that they can give rise to both embryonic and extraembryonic tissues. In contrast, cells of the ICM and TE become lineage-restricted: ICM cells are pluripotent, while TE cells are multipotent. These lineage properties are recapitulated by cell culture models, such as embryonic stem (ES) cells derived from the ICM and trophoblast stem cells (TSCs) derived from the TE.

Gene expression, 3D genome organization, and their regulatory factors vary across different cell states and potencies exhibiting heterogeneity between individual cells. This is particularly evident in early embryos, which represent a rapidly changing mosaic of distinct cellular states following fertilization. To capture these complex and heterogeneous processes – and to uncover the causal role of dark genome elements – we map changes in nuclear DNA, RNA, and protein composition as cell fate is progressively specified.
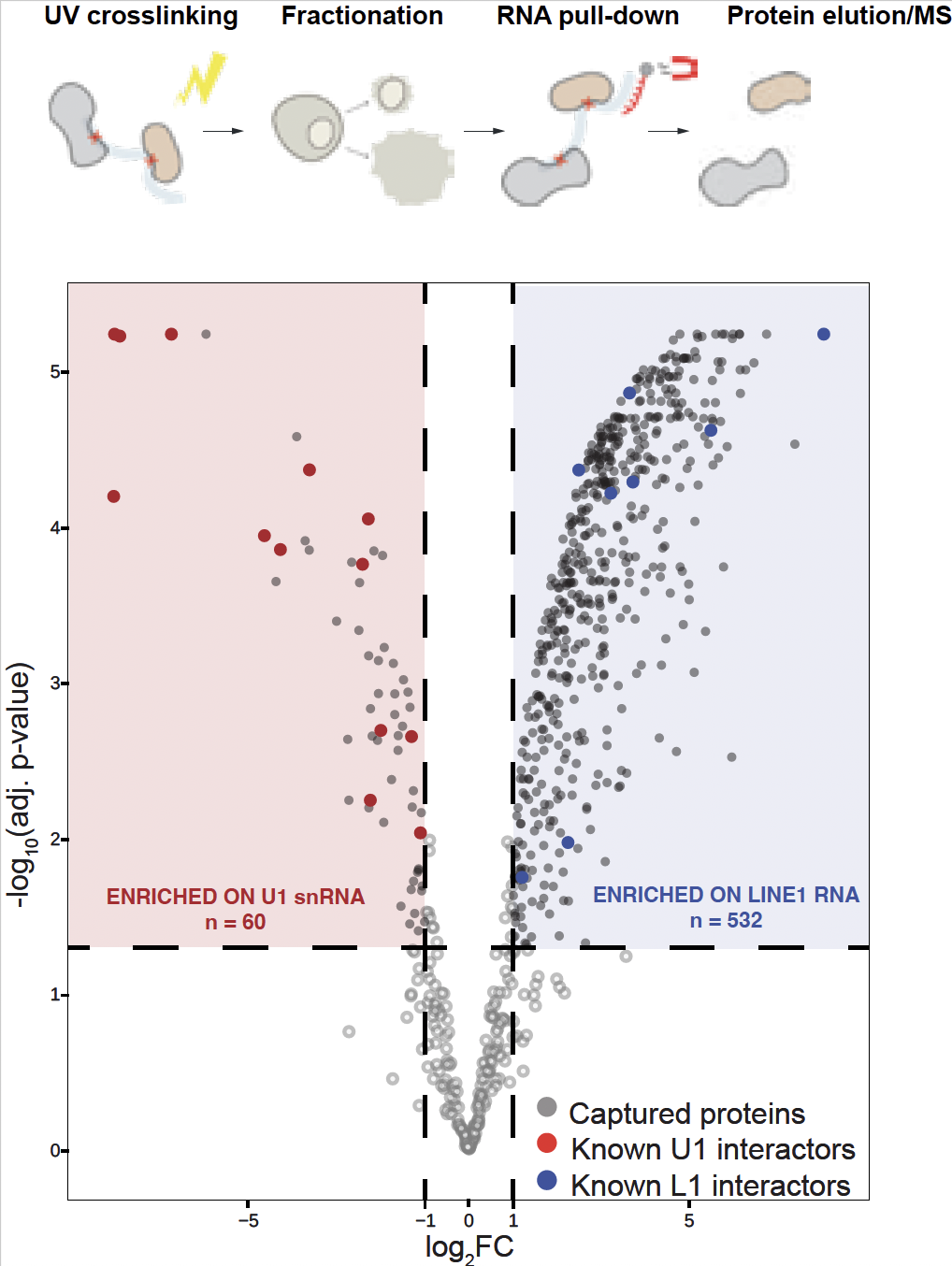
We employ and develop novel genome-wide sequencing methods to measure 3D genome interactions across scales, multi-omics spatial organization of DNA and RNA, and both RNA–protein and protein–RNA interactions including RAP-DNA, RAP-MS, CLAP/CLIP. For functional assays, we use large-scale CRISPR-based perturbations as well as targeted knockout and knockdown approaches, applied both in vivo and in cell culture. We integrate these perturbations with microscopy and sequencing-based techniques to comprehensively analyze molecular and spatial effects. We work closely with IMBA/IMP and Vienna BioCenter facilities including NGS platform, proteomics, molecular biology services, and Biooptics to optimize and further develop our methods.
One of the main techniques we use is Split-Pool Recognition of Interactions by Tag Extension (SPRITE) technology. SPRITE enables the measurement of higher-order DNA structures with high resolution for both short- and long-range regulatory elements in single cells. Because SPRITE technology is available as a multimaps measurement, it can detect various classes of RNAs (e.g., mRNAs, lncRNAs, small RNAs) and simultaneously capture 3D DNA structure alongside RNA levels and localization.