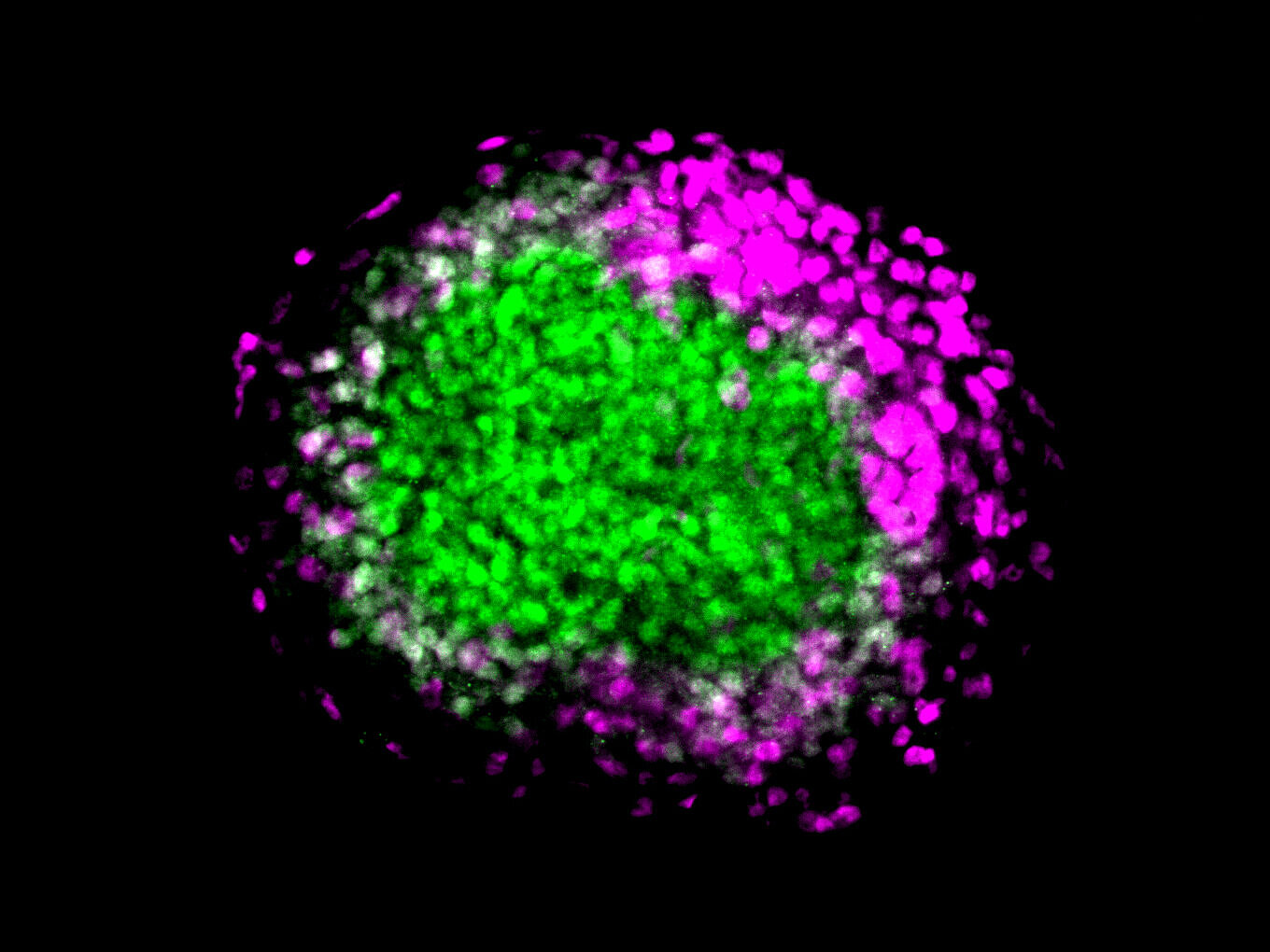
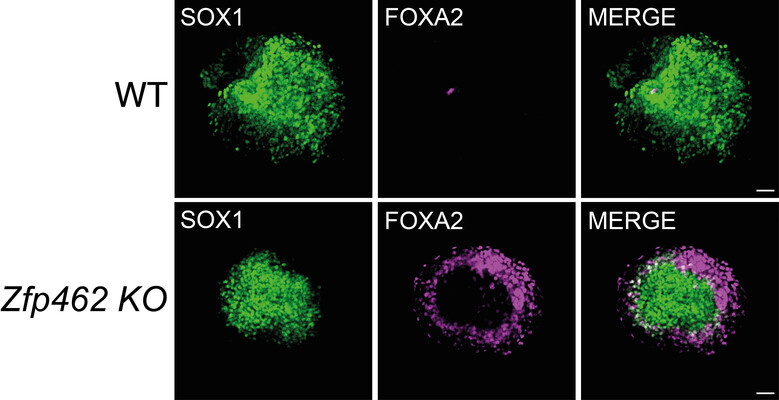
Immunofluorescence images of wildtype (WT) and Zfp462 deleted (Zfp462 KO) cells during neural differentiation. The neuronal lineage marker SOX1 is shown in green and the endodermal lineage marker FOXA2 is in magenta. Non-neural cells are detected during the neural differentiation of Zfp462 KO cells.
© Bell Lab / NCB /IMBA.
Mutations in genes encoding heterochromatin regulators are frequently associated with neurodevelopmental disorders (NDDs). Heterochromatin, tightly packed DNA, is thought to stabilize neural cell identity and function by enforcing transcriptional silencing of lineage non-specific genes through epigenetic chromatin modifications. However, most heterochromatin modifiers are ubiquitously expressed and lack sequence-specificity. Hence, it remains unclear how the precise targeting of gene silencing is controlled and how mutations in heterochromatin modifiers contribute to NDD-associated neuronal defects.
The researchers at IMBA and USC now uncovered the molecular mechanism that targets the heterochromatin regulators G9A/GLP to chromatin to repress lineage non-specific genes during early neurogenesis. They found that the previously uncharacterized murine zinc finger protein ZFP462 directly interacts with G9A/GLP. By doing this, ZFP462 establishes repressive heterochromatin at enhancers of mesoderm and endoderm-specific genes in mouse embryonic stem cells and neural progenitor cells. Failure to target G9A/GLP to non-neuronal enhancers in Zfp462 mutant cells resulted in their aberrant expression. This, in turn, led to the misspecification of neural cells towards meso- and endodermal cell lineages. In humans, a mutation on one allele of the orthologous ZNF462 gene causes a rare genetic disorder known as Weiss-Kruszka Syndrome and presents a high-confidence risk for autism spectrum disorder. Based on the results in mouse cells, it is plausible that the neuropathology in individuals suffering from Weiss-Kruszka Syndrome is caused by defects in neural cell specification.
“We demonstrated that ZFP462 plays a key role in safeguarding neural lineage specification in cells of early mouse embryos,” says first and co-corresponding author Ramesh Yelagandula, who started this project during his postdoc in the research group of former IMBA group leader Oliver Bell. In their study, Yelagandula, Bell, and their team showed that ZFP462-mediated recruitment of G9A/GLP facilitates the formation of heterochromatin at non-neural enhancers. Heterochromatin restricts the binding of activating transcription factors and prevents the expression of genes involved in specification towards mesodermal and endodermal cell lineages. Thus, ZFP462 is essential to promote ectoderm identity so that neuronal cells can develop properly.
“The three cell lineages leading to ectoderm, mesoderm, and endoderm emerge very early during embryonic development,” says last and co-corresponding author Bell. Ectodermal cells further differentiate into neuronal cells and tissues such as nerves, brain, and spinal cord. “Once the different cell lineages are specified, maintaining their identity is critical for safeguarding the function of particular cells and tissues,” says Yelagandula. Loss of cell identity can cause developmental defects and lead to disease. Therefore, effective epigenetic silencing by heterochromatin regulators is needed to maintain lineage specification and cell identity.
ZNF462, the gene implicated in Weiss-Kruszka syndrome, is highly conserved in vertebrates. By characterizing the mechanism of the mouse ortholog Zfp462, the new findings shed light on the possible molecular mechanism underlying a rare human neurodevelopmental disorder. “Our data suggest that Weiss-Kruszka syndrome could arise from the aberrant activation of lineage non-specific genes in the neuronal lineage during early embryogenesis,” says Bell.
The researchers are considering possible applications to help treat affected patients. Patients diagnosed with Weiss-Kruszka syndrome carry only one mutant allele of ZNF462 harboring an early stop codon, while the second allele is functional. As only one normal allele is present, the level of functional protein is reduced.“We are currently investigating if the mutant allele produces a truncated, potentially misfolded protein that has a dominant-negative role in ZFP462 regulation. If this were the case, we might be able to intervene by neutralizing the truncated protein and help alleviate the patients’ symptoms,” reports Yelagandula.