GMI and MFPL researchers develop a new method for rapidly identifying genes that contribute to pathogen virulence
23.04.2018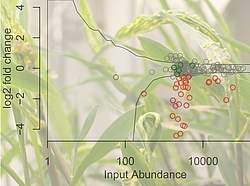
To combat pathogen infection in both animals and plants, it is important to know which genes in the pathogen are important for successfully infecting their host. Traditionally, identifying such genes has been laborious and time consuming. Researchers would mutate genes thought to be involved in infection, infect these mutants into their host one at a time, and then determine whether the mutant is as infectious as the non-mutated pathogen. In work published today in PLoS Biology from the Gregor Mendel Institute for Molecular Plant Biology of the Austrian Academy of Sciences and the Center for Integrative Bioinformatics Vienna at the Max F. Perutz Laboratories , both located at the Vienna BioCenter, PhD student Simon Uhse in the group of Armin Djamei describes a method that dramatically increases the speed for identifying genes critical for infection.
The new method, iPool-Seq, was developed to identify genes necessary for the fungus Ustilago maydis to infect its host Maize. The researchers made hundreds of individual mutants by inserting a specially marked DNA sequence into the fungi’s genes. They then mixed these mutants together in equal quantities, and infected maize with this pool of fungal mutants. According to Simon, “We reasoned that if we disrupted a gene important for infection, that mutant would not be present in the infected plant. The problem is, identifying the specific DNA sequences that mark each mutant is like looking for a needle in a haystack.”
To overcome this problem, they used a specialized form of PCR amplification to attach a chemical modification specifically to the DNA of the mutated fungus and separate it from the much more prevalent DNA of the Maize host. Using genome sequencing with specialized barcodes, they were then able to quantitatively determine which mutants were more or less infectious than the progenitor strain.
According to Armin Djamei “We were able to identify 23 new genes from U. maydis that are involved in infection, and which may be useful targets for combatting pathogen infection in the field. More importantly, we believe this technique could be used to rapidly and systematically identify key genes used by microbes to interact or infect not only plants but also animals, including humans.”